 校历
校历 邮箱
邮箱 EN
EN新一轮科技革命和产业变革加速演进,人工智能和生物信息学交叉领域重要科学问题和关键核心技术已经产生革命性突破,新的学科分支和产业新增长点不断涌现,学科深度交叉融合势不可挡。会议围绕人工智能技术对结构生物学研究范式和产业应用的影响和生物信息学前沿发展及其与人工智能新技术交叉融合趋势两个议题进行深入讨论。会议主题:学科交叉、融合创新、人才培养、产业转化。会议邀请国内人工智能、生物信息学、结构生物学、药学、医学等领域专家作专题报告,探索科技前沿。
一、会议组织:
主办单位:浙江省生物信息学学会人工智能专业委员会、浙江工业大学
承办单位:浙江工业大学信息工程学院
协办单位:中国生物信息学学会(筹)多组学与整合生物学专业委员会、生物大分子结构预测与模拟专业委员会
名誉主席:俞 立、陈 铭
会议主席:张贵军
组委会:张 彪、周晓根
秘书:刘 俊(电话:13372541308 邮箱:junl@zjut.edu.cn)
二、会议时间、地点:
时间:2022年11月12日
线上地点:腾讯会议(会议号:143-249-473)
线下地点:浙江工业大学信息工程学院D533会议室
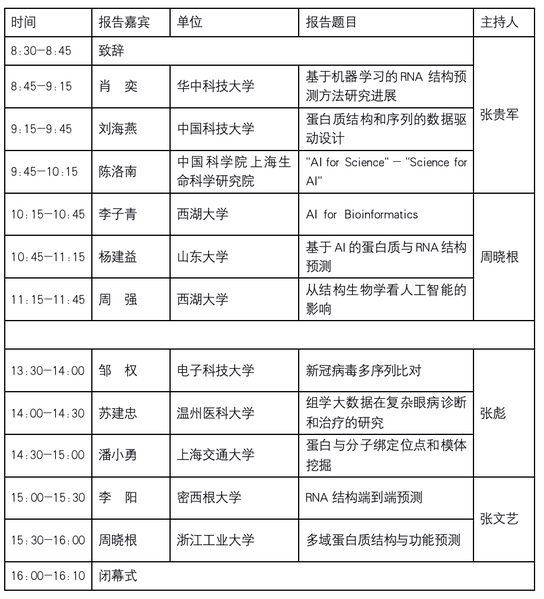
三、会议议程:
四、报告简介:
报告一:基于机器学习的RNA结构预测方法研究进展
内容简介:RNA分子参与各种生物过程,如催化和调控过程。为了完成这些功能,它们需要形成特定的结构。由于实验解析的RNA结构数目非常有限,人们发展了计算方法预测RNA结构。近年来,机器学习方法越来越多地应用于RNA结构预测,在许多方面预测准确性有了显著的提高,显示出很好的发展前景。这里将简要介绍RNA级结构预测研究的背景和现状,特别是基于机器学习的RNA结构预测方法方面的研究进展。

专家简介:肖奕 华中科技大学物理学院教授,国家杰出青年科学基金获得者。1981年获湖南师范学院物理系理学学士学位,1984年获中国科学院生物物理研究所理学硕士学位,1988年获上海交通大学应用物理系理学博士学位。《Biophysics Report》副主编,中国细胞生物学会功能基因组信息学与系统生物学学会副会长,主要研究方向为蛋白质和RNA折叠物理机制以及核酸单体与复合物结构预测、生物网络结构和动力学。
报告二:蛋白质结构和序列的数据驱动设计
内容简介:按需定向设计蛋白质等生物大分子具有重要理论和应用价值,也是极具挑战性的计算科学问题。深度学习等人工智能方法相对与传统计算设计方法的成功主要以其对高维复杂分布能进行更准确估计和更高效抽样为基础。目前相关方法进展集中于基于语言模型的氨基酸序列设计、基于结构的逆折叠、新主链结构从头设计等方面。我将介绍我们在蛋白质主链结构设计、氨基酸序列从头设计方面的进展,并讨论计算和实验指标对方法评估的价值。

专家简介:刘海燕 中国科学技术大学生命科学学院教授,国家杰出青年基金获得者,国家自然科学基金创新群体项目负责人。1990年本科毕业、1996年博士毕业于中国科学技术大学生物系。主要研究方向为蛋白质设计理论与实验方法及其应用、蛋白质空间结构和动力学的计算机模拟方法与应用。在蛋白质计算设计方面建立并实验验证了蛋白质结构序列从头设计的统计能量模型;在分子模拟方法方面发展了酶反应自由能面计算、集合自由度增强采样、单参考态自由能计算等技术;已发表研究论文百余篇。
报告三:AI for Science - Science for AI
内容简介:AI for Science: Spatially resolved transcriptomics (SRT) technology enables us to gain novel insights into tissue architecture and cell development, especially in tumors. However, lacking computational exploitation of biological contexts and multiview features severely hinders the elucidation of tissue heterogeneity. Here, we propose stMVC, a multi-view graph collaborative-learning model that integrates histology, gene expression, spatial location, and biological contexts in analyzing SRT data by attention. Specifically, stMVC adopting semisupervised graph attention autoencoder separately learns view-specific representations of histological-similarity-graph or spatial-location-graph, and then simultaneously integrates two-view graphs for robust representations through attention under semi-supervision of biological contexts. stMVC outperforms other tools in detecting tissue structure, inferring trajectory relationships, and denoising on benchmark slices of human cortex. Particularly, stMVC identifies disease-related cell-states and their transition cell-states in breast cancer study, which are further validated by the functional and survival analysis of independent clinical data. Those results demonstrate clinical and prognostic applications from SRT data.
Science for AI: Backpropagation (BP) algorithm is one of the most basic learning algorithms in deep learning. Although BP has been widely used, it still suffers from the problem of easily falling into the local minima due to its gradient dynamics. Inspired by the fact that the learning of real brains may exploit chaotic dynamics, we propose the chaotic backpropagation (CBP) algorithm by integrating the intrinsic chaos of real neurons into BP. By validating on multiple datasets (e.g. cifar10), we show that, for multilayer perception (MLP), CBP has significantly better abilities than those of BP and its variants in terms of optimization and generalization from both computational and theoretical viewpoints. Actually, CBP can be regarded as a general form of BP with global searching ability inspired by the chaotic learning process in the brain. Therefore, CBP not only has the potential of complementing or replacing BP in deep learning practice, but also provides a new way for understanding the learning process of the real brain.

专家简介:陈洛南 中科院生化细胞研究所研究员,中国科学院系统生物学重点实验室执行主任,国科大杭高院首席教授,国家重点研发项目首席科学家。华中科技大学电气工程学士学位;获日本东北大学系统科学硕士学位;获日本东北大学系统科学博士学位。1997年起任日本大阪产业大学副教授;2000年起任美国加州大学洛杉矶分校(UCLA)访问教授;2002年起任日本大阪产业大学教授。中国运筹学会《计算系统生物学分会》名誉理事长,IEEE SMC学会《系统生物学技术委员会》主席,中国生化细胞学会《分子系统生物学专业分会》主任委员。主要从事计算系统生物学、大数据分析和人工智能的研究工作。近年来发表400余篇期刊论文(包括 Nature, Nature Genetics, Nature Cancer, Nature Communications, PNAS, PRL, National Science Review, Cancer Cell等)和两部专著(H-index > 73 , Citation > 22000)。
报告四:AI for Bioinformatics
内容简介:介绍AI数据科学的流形原理,和最近AI+ 多组学数据分析、蛋白质结构预测、蛋白质序列设计的一些新进展。

专家简介:李子青 西湖大学人工智能讲席教授,IEEE Fellow,IAPR Fellow。曾任微软亚洲研究院Research Lead,模式识别国家重点实验室资深研究员。发表论文500余篇,撰写编写著作10部,Google Scholar引用超过56700。曾任AI顶刊IEEE T-PAMI等刊物副主编,担任100余个国际学术会议大会主席、程序主席或程序委员。作为机器学习和计算机视觉专家,在微软领导开发的人脸识别系统 EyeCU,比尔.盖茨接受CNN采访为之讲解;代表中国国家体撰写了中国第一个ISO/SC37生物识别国际标准工作草案并获采纳。2019年加入西湖大学,现研究方向为(1)机器学习、深度学习、数据科学基础理论与方法,(2)AI+学科交叉应用(计算机视觉、生命科学、生物医学、环境科学,等)。在研基金委重点项目(AI+多组学分析)项目负责人,科技部“新一代人工智能”重大项目项目(AI+蛋白质计算、药物设计)项目负责人/首席科学家。立足科技前沿,深入科研一线,指导、培养了众多杰出学生,部分成为AI学科教授、院长,或国内外知名AI企业CEO、CTO。
报告五:基于AI的蛋白质与RNA结构预测
内容简介:AlphaFold2等方法使蛋白质结构预测取得了突破性进展。课题组前期开发了蛋白质结构预测方法trRosetta,在AlphaFold2的启发下,我们对trRosetta做了一系列发展,本报告将对它们做简要介绍。

专家简介:杨建益 山东大学教授、博士生导师,国家杰出青年基金获得者。曾就职于南开大学、密歇根大学和华盛顿大学。与合作者共同开发了trRosetta和I-TASSER等著名蛋白质结构预测算法,在Nature Methods、PNAS等发表论文60余篇,论文被SCI他引8000余次。更多信息请见课题组网站:http://yanglab.qd.sdu.edu.cn/
报告六:从结构生物学看人工智能的影响
内容简介:结构生物学是利用物理学手段将生物学建筑在化学基础上的一门学科。结构生物学是生物学的重要领域,它使得生物学家可以从化学角度分析生物学问题。长期以来,结构生物学一直是一门以实验为主的科学。近年来,人工智能技术引领革命性突破,推动新一轮科技革命加速演进。人工智能赋予了结构生物学新的动能,促使其从新的视角和新的水平上研究和看待问题。报告将围绕会议主题探讨人工智能技术对结构生物学研究范式的改变。

专家简介:周强 西湖大学生命科学学院特聘研究员、博士生导师,教育部“长江学者奖励计划”特岗学者,国家优秀青年科学基金项目获得者。周强研究员长期从事生物大分子冷冻电镜的研究,结合多种手段研究与重大疾病或者重要生物学过程相关的膜蛋白的结构以及工作机制,在新冠病毒入侵细胞机制、人源溶质转运蛋白等研究方面取得了一系列成果。周强课题组率先解析了新冠病毒受体ACE2与新冠病毒S蛋白复合物的三维结构,揭示了新冠病毒入侵人体第一刻的情形,为理解新冠病毒入侵细胞的机制以及开发针对性的药物、抗体和疫苗提供了结构基础。周强获得了多个荣誉奖项,包括教育部“长江学者奖励计划”特岗学者、浙江省自然科学一等奖(第一完成人)、药明康德生命化学研究奖、国家自然科学基金委优秀青年科学基金项目负责人、中国电子显微镜学会优秀青年学者奖,浙江省“万人计划”青年拔尖人才、浙江省抗击新冠肺炎疫情先进个人、杭州市五一劳动奖章、杭州市十大青年科技英才、杭州市领军型创新创业团队负责人等。周强研究员作为通讯作者在Science、Nature、Cell、Cell Research、Science Advances、Nature Communications、Cell Discovery等杂志上发表20多篇文章。目前主持国家自然科学基金委优秀青年科学基金和1项面上项目,作为子课题负责人参与科技部重点研发计划1项,完成浙江省重点研发计划1项。
报告七:新冠病毒多序列比对
内容简介:目前已从世界各地发现并测序了接近100万条新冠病毒DNA序列,新冠病毒序列高度保守但存在多处有语义和功能差异的变异。对新冠病毒序列进行多序列比对有助于高效地发现变异位点、重现进化关系。但是鉴于新冠病毒序列数目多、序列长度长,目前主要的多序列比对软件无法完成。我们的前期工作HAlign可以快速处理大规模相似DNA序列,有望解决该问题。在比对新冠DNA序列的过程中,我们进一步的优化HAlign,包括:首位序列的处理、部分插入空格的优化、以及后缀树的数据结构设计等方面。优化后的HAlign在比对效果和比对时间上都优于目前现有的软件,比对时间缩小了十倍以上。通过对新冠病毒序列全面的多序列比对,我们还分析出若干新知识。

专家简介:邹权 电子科技大学教授、博士生导师,国家优秀青年基金获得者。2009年于哈尔滨工业大学计算机学院获得博士学位。2018年调入电子科技大学基础与前沿研究院。主要研究方向为生物信息学、机器学习和字符串算法。目前担任SCI期刊Current Bioinformatics主编,IEEE Access、Frontiers in Genetics、Frontiers in Plant Science副编辑和5个SCI期刊的编委,获授权国家发明专利一项;入选科睿唯安2018-2020年全球高被引学者;四川省特聘专家(四川省千人计划);2019年获得国家自然科学基金优秀青年基金资助;2019年获教育部自然科学二等奖(排名第二);2020年获福建省自然科学三等奖(排名第一)和吴文俊自然科学奖(排名第二);其中代表作发表在Bioinformatics、PLOS Computational Biology、RNA等知名学术期刊上。相关论文被多篇Nature子刊引用;率先采用MapReduce并行框架和字符串算法突破了多序列比对难题的计算瓶颈,相关软件被美国、欧洲、印度科学院院士高度评价,并受到中科院官网、新浪科技等媒体报导;提出的集成分类算法不但是学术期刊Neurocomputing官网下载次数最多的热点论文之一,而且得到产业化应用,用于百度贴吧的反作弊系统,受到百度主题研究项目资助和百度公司官方报导。
报告八:组学大数据在复杂眼病诊断和治疗的研究
内容简介:据世卫组织统计,全球至少有22亿人存在视力障碍或失明,我国是致盲人数最多国家。其中至少有10亿人存在视力障碍,这种情况本可以预防或尚未得到治疗。眼部疾病非常普遍,那些活得足够长的人一生中至少会有一种眼部疾病。本报告将从组学大数据角度汇报课题组近期在近视预警和防控,高度近视、青光眼、糖网等多种致盲性眼病的遗传模式和调控机制,及潜在基因治疗途径等方面的进展。

专家简介:苏建忠 温州医科大学教授、博士生导师,国家高层次引进海外人才。温州医科大学生物医学大数据研究所所长,浙江省高校领军人才,浙江省杰出青年基金获得者。主持国家自然科学面上项目、国家自然科学重点基金子课题,科技部重点研发子项目等科研课题。主要从事眼病遗传诊断,复杂疾病的遗传和表观遗传调控模式,单细胞表观基因组检测技术和模式识别算法开发等方面的研究工作。开发在线算法工具和数据库10余个,发表高水平SCI论文80余篇,其中第一或通讯作者(含共同)在Nature Genetics、Blood、Nature Communications、Genome Biology、Nucleic Acids Research、PNAS及 ophthalmology 及IOVS 等国际高水平期刊发表研究论文50余篇,获省部级科研奖项2项。担任浙江省生物信息学会健康医学和转化医学委员会主任委员,中国卫生信息与医疗健康学会眼视光专委会委员,中华预防医学会生物信息学委员,浙江省生物信息学会理事。
报告九:蛋白与分子绑定位点和模体挖掘
内容简介:蛋白与RNA、化合物分子相互作用形成的网络是认识生命系统复杂现象的重要内容。DNA元件百科全书(Encyclopedia of DNA Elements, ENCODE)项目绘制了150多种RNA绑定蛋白的结合与功能图谱。这些数据使得我们能够开发机器学习模型挖掘RNA、小分子和蛋白绑定的序列与结构特征。我们考虑绑定位点的序列和结构上下文、全局和局部特性,开发了一系列的基于深度网络模型的绑定位点和模体预测方法。

专家简介:潘小勇 上海交通大学自动化系长聘教轨助理教授、博士生导师。博士毕业于丹麦哥本哈根大学,随后在荷兰伊拉斯姆斯医学中心和根特大学从事博士后学习。主要从事模式识别与生物信息研究,在Nature Biomedical Engineering, Bioinformatics、NAR等期刊上发表多篇论文。
报告十:RNA结构端到端预测
内容简介:RNA是细胞的重要基础,通过形成特定三维结构来执行相应的功能。从序列直接预测RNA的三维结构是一项具有挑战性的任务。该报告将介绍一种新的基于深度学习的RNA三维结构预测方法。该方法实现了RNA结构和远程核苷酸之间的几何相互作用的端到端预测,通过将预测的约束信息转变为混合势能,指导后续的构象优化过程。相比传统RNA结构预测算法,该算法具有一定的优势。

专家简介:李阳 博士,2014年获得南京理工大学计算机科学与工程专业的学士学位,2021年获得南京理工大学控制科学与工程的博士学位,2017年至2022年于密西根大学张阳实验室进行联合培养。主要研究方向为结构生物信息学。在CASP13和CASP14的残基接触预测赛道中分别排名第一和第二。
报告十一:多域蛋白质结构与功能预测
内容简介:蛋白质结构预测是阐明功能机理和揭示生命体生物学本质的基础,是加速疾病研究和促进创新药物研发、疫苗设计和精准诊断的核心技术。AI蛋白质结构预测程序AlphaFold2实现了基于氨基酸序列精确预测蛋白质三级结构的重大突破。AlphaFold2虽然精度较高,但多域蛋白质的全长结构建模精度仍需进一步提高,且结构域缔结与协作实现的功能至关重要。本报告介绍多域蛋白质结构组装和功能预测方面的工作,包括基于类似模板的多域蛋白质组装方法、冷冻电镜密度图辅助的多域蛋白质结构渐进组装方法、全自动化的多域蛋白质结构和功能深度学习预测平台。

专家简介:周晓根 博士,浙江工业大学运河青年学者特聘教授。2018年毕业于浙江工业大学,2018至2021年于密西根大学张阳实验室从事博士后研究工作,2022年加入浙江工业大学。研究方向为结构生物信息学、计算智能及深度学习。第一作者在PNAS,Nature Computational Science、Nature Protocols、Nucleic Acids Research、Bioinformatics、TEVC、TCYB等期刊发表论文14篇;获得浙江省优秀博士学位论文奖;合作开发了DEMO、DEMO-EM和I-TASSER-MTD多域蛋白质结构建模和功能预测服务器;目前承担和参与国家自然科学基金项目、国家重点研发项目课题3项。

 浙公网安备 33010302002621号
浙公网安备 33010302002621号